Willkommen im BioNTech Medienbereich
In diesem Bereich finden Sie eine Vielzahl von Medieninhalten und nützlichen Informationen zum Herunterladen.
Medien-Downloads
Konferenz "Zusammenarbeit zur Förderung einer gerechten Impfstoffversorgung in Afrika", 18. Dezember 2023

























Einweihung der mRNA-Produktionsstätte in Kigali, Ruanda




















Marburg Produktionsanlage








mRNA-Produktionsstätte in Afrika































COVID-19








BioNTainer















,%20Özlem%20Türeci%20(CMO%20BioNTech),%20H.E.%20President%20Macky%20Sall%20(President%20of%20the%20African%20Union,%20President%20of%20Senegal),%20H.jpg)
,%20Özlem%20Türeci%20(CMO%20BioNTech),%20H.E.%20President%20Paul%20Kagame%20of%20Rwanda%20and%20Holm%20Keller%20(Chairman%20kENUP%20Foundation).jpg)

%20and%20Özlem%20Türeci%20(CMO%20BioNTech)%20speaking.jpg)
%20speaking.jpg)
%20speaking.jpg)
%20speaking%20(1).jpg)
%20and%20Özlem%20Türeci%20(CMO%20BioNTech).jpg)
%20speaking.jpg)
%20presenting.jpg)
%20speaking.jpg)

%20speaking.jpg)
%20speaking.jpg)
%20speaking.jpg)
%20speaking.jpg)

.jpg)
.jpg)
.jpg)
%20speaking_2.jpg)
%20speaking.jpg)



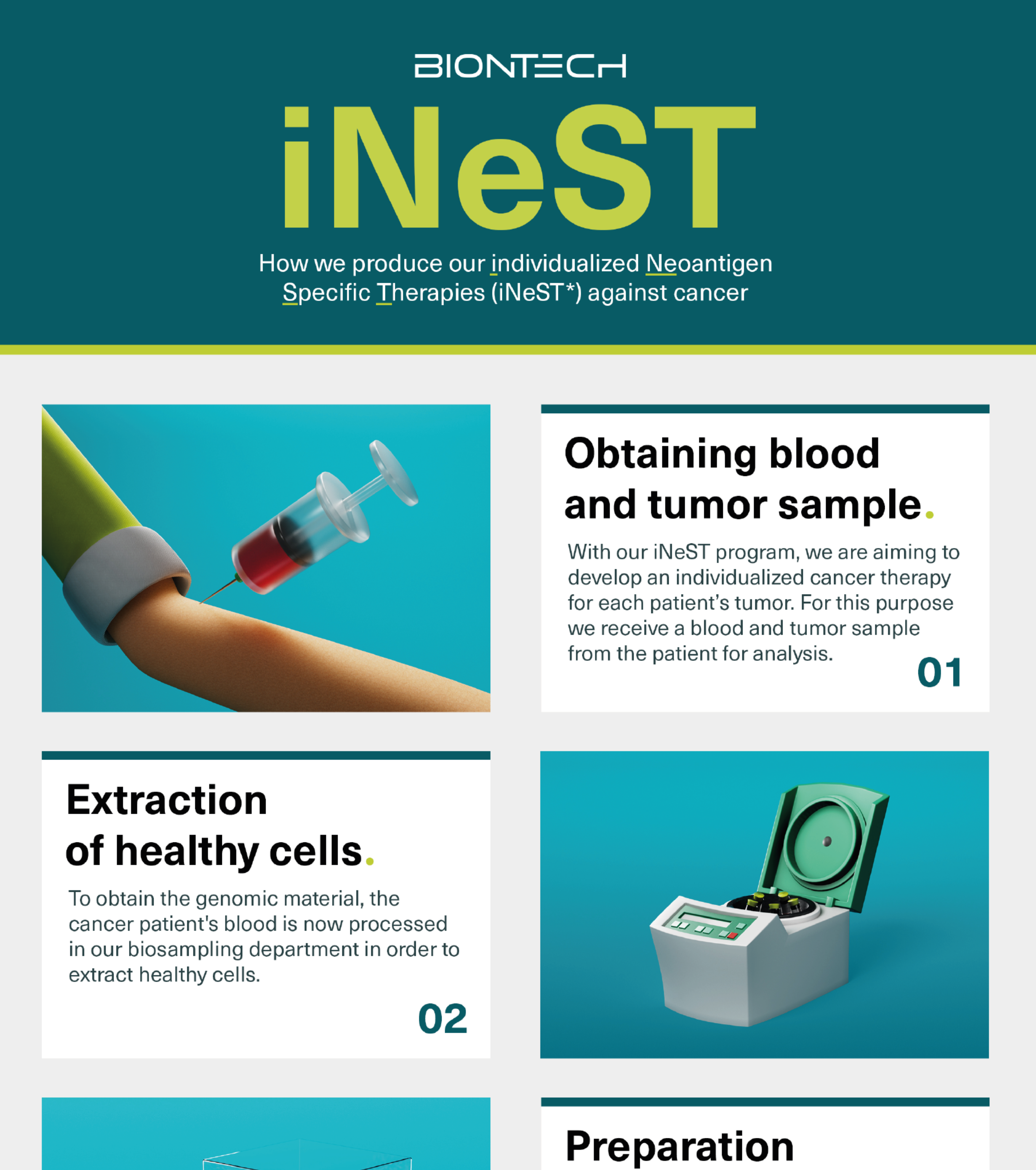
iNeST - individualisierte Krebsmedizin




%20jpg.jpg)
%20jpg.jpg)
.PNG)

.PNG)


.jpg)









Labore








Management







Aufsichtsrat






Standorte


Fact Sheets

BioNTech Logo

Alle Urheber- und Verbreitungsrechte an den Bildern auf dieser Website liegen bei der BioNTech SE. Die Verwendung von Bildern bedarf der Angabe der Quelle "© BioNTech SE 2023, alle Rechte vorbehalten". Bitte informieren Sie uns über die Verwendung von Bildern aus unserer Bibliothek vor der Veröffentlichung per E-Mail an media@biontech.de
Bitte beachten Sie, dass die Bilder nicht verändert oder in irgendeiner Weise mit anderen Bildern kombiniert werden dürfen. Eine dauerhafte Speicherung auf elektronischen Systemen ist nicht gestattet.
Weitere Informationen

Presseanfragen
Jasmina Alatovic
Vice President Corporate Communications
+49 (0)6131 9084-1513


